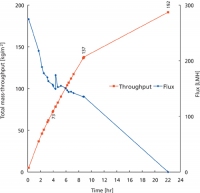
Figure 1: mAb total mass-throughput and volumetric flux through the Viresolve Pro filter as a function of time. An X0HC and a Viresolve Pro prefilter were used with 93 g/L mAb in 3 mM histidine, pH 5.0 at 35 °C. Newer classes of biotherapies will require innovations in processing technology.
By Glen R. Bolton, Bernard N. Violand, Richard S. Wright, Shujun Sun, Khurram M. Sunasara, Kathleen Watson, Johnathan L. Coffman, Christopher Gallo, and Ranga Godavarti
ABSTRACT: In recent years, most pharmaceutical companies have focused on the development of monoclonal antibodies (mAbs). Increasing upstream titers and shrinking development timelines have posed several challenges to downstream process development of mAbs. Some of the major strategies and tools to address these challenges include the development of highly efficient platforms, high-throughput screening (HTS) tools, and reduction of the number of unit operations to help with facility fit. In the future, mAbs may represent a smaller percentage of the pipeline as portfolios concentrate more on development of antibody fragments, nanobodies, biosimilar protein therapeutics, conjugated proteins and vaccines, Fc-fusions, and nonantibody protein scaffolds. In addition to high cell density mammalian expression, expanded utilization of other expression systems such as microbial and yeast will support these newer biotherapeutics (BioTx). The diversity of BioTx and expression systems will pose unique challenges for downstream development. Novel tools, approaches, and/or platforms will be required to enable rapid development. Furthermore, with the increasing emphasis on Quality by Design (QbD), there is a need to develop paradigms to apply QbD not only to mAbs but also to other BioTx.
Early biotechnology products in the US and Europe were characterized by low titers and low cell densities using various cell hosts (1, 2). Most companies had few recombinant products licensed or in development, including insulin, somatotropin, interferon, tissue plasminogen activator, erythropoietin, Factor VIII, and Factor IX. There were no benefits or need for platform development or expanding manufacturing capabilities. Cell culture was performed using numerous methods, including roller bottles and stirred tank bioreactors operated in perfusion, batch re-feed, or fed-batch mode (1).
Starting in 1986 with the licensure of Orthoclone Okt3 (muromonab-CD3), monoclonal antibodies (mAbs) such as Rituxan (rituximab), Herceptin (Trastuzumab), and Remicade (infliximab) have dominated the BioTx market (3). Most company pipelines saw a dramatic increase in the number of therapeutic mAb candidates, which in turn resulted in a desire to shorten development timelines. Due to the large doses required, antibodies are typically expressed at high titers in high cell density mammalian cell culture processes. This past decade has seen both the titers and cell densities increase by an order of magnitude to meet the clinical and commercial demands (4–6). Further, companies have made significant investments in manufacturing facilities, resulting in cell culture bioreactor capacities of 12,000–20,000L. The increasing volumes, cell densities, and protein masses have posed numerous new challenges for downstream processing.
Addressing Challenges in mAb Processing
Shrinking timelines have necessitated developing tools for faster process development. Because of the similarity of mAbs, which mainly differ from each other in their complementarity determining regions (CDR), these biomolecules are amenable to platform development and manufacturing processes. The standardization of cellular expression systems, bioreactor conditions, purification processes, manufacturing hardware, and disposables has resulted in faster clinical development and lower costs. Most companies have developed standardized purification processes typically consisting of a cell harvest method followed by a capture chromatographic step and one or polishing steps. The approach used at Pfizer has been described elsewhere (7–12). Weak partitioning chromatography (WPC) and a high throughput screening (HTS) tools have been essential for development of a robust two-column platform purification process. The benefits of a two-column purification process include reduced capital, footprint, quality systems, validation, cleaning, development costs, solutions, and water consumption. The HTS method has also been applied to the development of other purification processes for unique biotherapeutic modalities where screening of a large number of resins may be required.
Another consideration in mAb processing is reducing the cost of goods (COGs). The cost of the virus-retaining filter, which can approach that of the protein A resin, can be reduced by using prefilters or optimizing protein concentration, temperature, buffer composition, and solution pH (see Figure 1) (13). The costs of freezing and storing bulk drug substance can be reduced by targeting high concentrations. Concentrations above 250 g/L have been achieved using ultrafiltration at elevated temperatures (14) and over 500 g/L using a wet-ultrafiltration membrane evaporation method (15) (see Figure 2).
A significant consequence of the transfer of products between facilities is the generation of in-process pools that exceed the capacity of the storage vessels. Linking unit operations through tandem processes would eliminate this constraint and additionally may reduce process time, process costs, and documentation requirements. Feasibility has been demonstrated for a tandem downstream process for the purification of mAbs employing an affinity Protein A capture step followed by a flow-through anion-exchange (AEX) step and a virus filtration step (VRF) (16).
Future Challenges in Downstream Processing
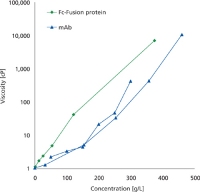
Figure 2: Viscosity versus concentration for an Fc-fusion protein and a mAb concentrated using a wet-ultrafiltration membrane evaporation method.
Because of the broad range of new molecules, hosts, and delivery methods being investigated, recent biopharmaceutical development has resembled that from the early stages of the industry. A broad range of alternative BioTx modalities have obtained regulatory approval and are currently in clinical trials (17–21) including antibody fragments, single-domain mAbs, Fc-fusions, vaccines, antibody-drug conjugates, nonantibody protein scaffolds, PEGylated proteins and peptides, and viral vectors.
Some of the newer BioTx use nonmammalian expression systems such as yeast and E. coli if there is no requirement for post-translational modifications. These alternate expression systems may offer cost advantages. However, other issues may arise, including undesired mannosylation in a transferrin-exendin-4 fusion molecule expressed in yeast (22). This unwanted modification required an extra process step using hydrophobic interaction chromatography (HIC) to separate these modified molecules from the desired product. E. coli expression often yields high levels of protein production, often in the form of inclusion bodies, which require extra steps involving their isolation and the subsequent extraction and refolding to obtain the desired product (23).
For a difficult-to-refold protein such as neurotrophin-4, an additional step involving sulfonation of the cysteines was also required to obtain a useful refold yield (24). The use of high throughput screening with Design of Experiments has been used to accelerate the determination of optimal conditions for refolding difficult proteins (25).
Each of these BioTx modalities poses a unique challenge in downstream processing because of the lack of a platform process resembling that used for mAbs. Examples of the purification challenges involved with two Fc-fusion proteins will be described in the following sections.
Because each Fc-fusion protein has a different protein sequence fused to an antibody Fc region, the protein amino acid sequence, charge, size, and hydrophobicity vary more than those of antibodies. In addition, they are often thermally, chemically, or enzymatically unstable, which can lead to high levels of aggregates, clips, or inactive species.
Fc-Fusion Proteins
Case study 1.
A purification process was developed for an acidic Fc-fusion protein that was unstable at ambient temperatures and low pH and that quickly formed high molecular weight oligomers (HMW1) and dimers (HMW2).
To improve stability, the protein A chromatography step was performed at 2–8 o C and the low pH eluate was neutralized immediately. The capacity of the protein A resin for the molecule was less than 15 mg/mL which is typical for Fc-fusion proteins.
A combination of HTS methods and gradient chromatography was employed to identify resins and operating conditions for high molecular weight (HMW) species removal. Anion exchange, cation exchange, and HIC resins were screened in 96 well plates in an HTS format. Additionally, ceramic hydroxyapatite (CHA), immobilized metal affinity chromatography (IMAC), and HIC methods were screened with elution gradients.
Due to the low pI of this Fc-fusion protein, a WPC–AEX step was not successful. Based on the resin screening, CHA chromatography was selected for optimization. An initial test of CHA was performed using a phosphate gradient. This method allowed identification of step elution conditions that reduced the HMW levels from 29% to 10.1% with 87% yield. The HMW1 flowed through the column and the peak contained primarily dimer species (HMW2). The CHA step had a capacity of 20 g/L.
The HTS analyses also indicated that a HIC resin under certain loading and elution conditions could provide reduction of HMW. An initial experiment using protein A peak pool and a salt gradient indicated the resin had a relatively low capacity for the product but was effective at reducing both the levels HMW species and less active monomer variants.
Further development was performed using the protein A pool as a load to the HIC step to identify conditions where the product would flow through the column while the HMW and less active monomer would bind. A successful method was developed but the HMW1 species began to flow through the column at about 10 mg/mL of load.
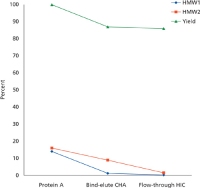
Figure 3: Yield and percentages of HMW1 and HMW2 versus step in the Fc-fusion purification process described in case study 1.
Because the CHA step was successful at removing the HMW1, but not HMW2 species, the CHA pool was used as a load for the HIC step. The HIC step was loaded with the CHA pool containing primarily HMW2 at 20 mg/ml. The HIC step provided a six-fold reduction in HMW2 levels. In addition, the HIC step reduced the level of host cell proteins by 1.4 log10 . Based on these data, the final process used a Protein A capture step with elution at low pH followed by immediate neutralization. This was followed by a bind-elute CHA step to remove HMW1 followed by a flow through HIC step to remove HMW2 (see Figure 3). The first two chromatography steps were performed in the cold to maintain product stability.
Case study 2.
During development of an early-phase purification process for an acidic Fc-fusion protein expressed in a CHO cell culture system, it was determined that the protein displayed a range of 24–100% activity. Several types of chromatographic resins, including cation exchange, anion exchange, HIC, CHA, and IMAC, were evaluated to remove the less active molecules along with other product and process-related impurities. The process and conditions described above for Fc-fusion protein 1 were not applicable to Fc fusion protein 2.
The CHA is a bimodal resin with two primary functional groups: phosphate and Ca2+ . The negatively charged phosphate groups serve as cation exchangers and the Ca2+ groups serve as chelators to proteins. For purification of basic and neutral proteins, the CHA column is usually equilibrated with phosphate buffer at a neutral pH to utilize a cation exchange mechanism for the separation of protein monomers from HMW species.
The CHA column, when equilibrated with phosphate, resolved fully active protein from partially active protein. The product was in the unbound fraction while the less active molecules were bound to the column. However, the load capacity was low and removal of high molecular weight species was poor using the phosphate-charged CHA column in flow-through mode.
A calcium-charged CHA column operated in bind-elute mode successfully removed the less active monomer species, as well as high molecular weight species, from the process stream. In this case, the CHA column was equilibrated with a low concentration of CaCl2 in a neutral pH buffer, and then loaded with a protein mixture also containing CaCl2. After washing the column with a neutral pH buffer, the column was eluted with a phosphate buffer and stripped with a high phosphate buffer. A high yield of the product was obtained in the elution peak. The less active protein and HMW species were present in the strip fraction. These data demonstrate that although Fc-fusion proteins are more challenging to purify than mAbs, other chromatography resins can be used to yield high-quality product.
These two examples illustrate the challenges posed by the different Fc-fusion molecules. In one case, calcium charging of the CHA column was required for removal of a lower activity monomer species. In another case, a bind-elute CHA column followed by a flow through HIC column was required for adequate HMW removal, resin capacity, and product yield. The sequence of the polishing steps is also important for both purity and capacity.
Antibody-Drug Conjugates
Numerous antibody drug conjugates (ADCs) are in clinical trials with most of them being used in cancer treatment with a toxin payload (26). The major downstream challenge is separation of multiple species containing different levels of the attached drug to the mAb. This results from the limited specificity for chemical conjugation of the drug to either exposed lysines or cysteines (generated by limited reduction) on the mAb. To enable straightforward downstream processing of ADCs, two main strategies have been used to generate more homogeneous molecules. These are the incorporation of additional free cysteines that can then be chemically targeted specifically or to mutate out some of the native cysteines to serines so that they are less available for coupling after a limited reduction reaction (27, 28).
mAB Fragments and Single Domain mAbs
One approach to generate smaller and simpler BioTx for downstream processing is to replace full-length mAbs with antibody fragments such as single-chain variable fragments (scFvs) and antigen-binding fragments (Fabs). Cimzia and Lucentis are two recently approved Fabs produced in E. coli (29) and although no scFv has yet received regulatory approved, there are a large number of both types of Ab fragments in clinical trials (30–32). Purification of these Ab fragments utilizes normal chromatographic procedures unless the H-chain is from the VH3 IgG gene family in which case it will bind to protein A. After purification these small fragments are usually modified to improve their pharmacokinetics using technologies, such as PEGylation. PEGylation dramatically changes the chromatographic properties of proteins such that loading capacity is severely compromised thus requiring larger columns (33, 34). Additionally PEGylation can dramatically increase the viscosity of concentrated drug substances.
In addition to mAb fragments, another approach to obtain smaller less complex antibody molecules is to use either Domain Abs (human derived) or nanobodies (Llama derived), which are composed of a single variable domain of ~ 14kDa. These small single domain antibodies can be easily captured from fermentation broth using Protein A since they can be derived from the VH3 IgG gene family (35, 36). Subsequent downstream purification has been accomplished using standard chromatographic techniques (36).
Viral Vectors
Using viral vectors as BioTx for gene therapy offers many exciting opportunities to inactivate new targets. Their production presents numerous challenges because of the complex nature of their composition and large size (2–5 x 106 kDa). They are usually composed of multiple proteins that encapsulate the nucleic acid payload. Additionally, there may be empty capsids that do not contain the gene of interest but are similar in physicochemical properties and can be difficult to separate from the desired product. CsCl density gradient centrifugation has long been the method of choice for their isolation; however, for clinical testing, larger scale production has recently been accomplished using standard column chromatographic methods. However, because of their large size, their binding capacity is usually very low and chromatographic media with normal pore sizes used for proteins offer no advantages since the pores are not available. Smaller beads with more surface area is the most effective manner to increase capacity (37,38). Other processes that are being utilized include tangential flow filtration that takes advantage of the large particle size for separation from smaller cellular contaminants.
Vaccines
Vaccines can be produced using numerous technologies and have been generated against proteins, polysaccharide, and small molecule targets. Pneumonia is the largest single infectious disease causing infant mortality (39). To date, three vaccines have been licensed for treating diseases caused by pneumococcus-containing polysaccharides conjugated to proteins (40).
Because prophylactic vaccines are given to a large healthy population ranging from infants to the elderly, the regulatory requirements during development and for licensure tend to be more stringent as compared with other biotherapeutic products. This also results in more early investment and upfront development work for a vaccine candidate than for a model biotherapeutic agent. Most commercial prophylactic vaccines are not well-characterized biologicals from a regulatory perspective because of their inherent complexity and/or poorly understood mechanism of action. In addition, if an in vivo animal potency model is available, the results don’t usually predict the human response to the product with regards to the protein structure-immunogenecity/antigencity relationship. These reasons force sponsors to initiate extensive process characterization studies and to lock the production processes early in the clinical development program (41).
One new approach to generate vaccines is to use virus–like particles (VLP) as potent immunostimulaters with covalent attachment of many copies of the desired antigen to their surface (42). Similar to viral vectors, VLPs present unique challenges because of their large size (2.5 x106 kDa). Q (14 kDa) is the most commonly used VLP. It is derived from the structural coat protein of this virus and it naturally assembles into 180 copies of itself during expression in the cytoplasm of E. coli. Subsequent purification is accomplished by classical column chromatography techniques with size-exclusion chromatography cited most frequently, which is not a desired step for scale-up (41). The challenges to develop alternative process steps are similar to the viral vector BioTx because of low capacity for most resins as well as the fact that there may be heterogeneous levels of antigen attached to the VLPs. Another new approach is the delivery of plasmid DNA to cells, which leads to the transcription of antigens and a subsequent immune response (43).
An additional complexity is the fact that many vaccines are multivalent. The manufacturing processes for these vaccines involve many different fermentation, purification, and conjugation trains for the polysaccharides and carrier proteins. Developing processes that are similar between antigens can improve manufacturability and facility fit (44).
Biosimilars
The FDA and European Medicines Agency (EMA) have indicated that the clinical testing requirements for a biosimilar drug can be reduced if it can be demonstrated that the biosimilar candidate does not meaningfully differ from the innovator drug (45, 46). Currently developed biosimilars, such as human growth hormone, insulin, and erythropoietin, are chemically far simpler than mAbs. It is unlikely that blood or plasma-derived products or complex vaccines will be acceptable as biosimilars in the US or Europe due to their complexity (47). However, eight categories of biosimilar molecules have been manufactured in India, including monoclonal antibodies and recombinant hepatitis B vaccine (48). The first monoclonal antibodies that will lose patent protection in Europe and the US were made using more stringent impurity level targets than exist now because the technology is more mature and the safety and efficacy is well established. For example, early mAbs had levels of high molecular weight species below 0.5% (49). It is possible that mAbs in the future will target levels below 5% (50).
A new purification challenge will be to produce antibodies with product quality attributes that match the innovator antibodies. This may be carried out with a host cell and/or purification process that differs from the innovators. A biosimilar antibody can have different levels of impurity or product variant species than the innovator, if it can be justified. The biosimilar manufacturer will determine the amount of clinical and characterization data that will be used to justify differences to the agencies. Therefore each biosimilar manufacturer must balance the development costs required to match innovator antibody characteristics against the costs of justifying differences. During development of biosimilar recombinant human growth hormone, higher levels of host cell proteins lead to increased levels of antibodies to both host cell proteins and growth hormone. This necessitated modifications to the purification process, which eventually allowed demonstration of comparability to the innovator and approval by the EMA (51). The success of a biosimilar development program will benefit from robust, selective, and efficient purification processes that require minimal development time and material.
Implementing Quality by Design
The past decade has witnessed an increasing emphasis—by both industry and regulatory agencies—on implementing Quality by Design (QbD) principles. In 2008, the FDA’s Office of Biotechnology Products invited companies to participate in a pilot program involving the submission of quality information for biotechnology products in an Expanded Change Protocol (52,53). The purpose of the pilot program is “to gain more information on and facilitate agency review of QbD, risk-based approaches for manufacturing biotechnology products”. These approaches link “attributes and processes to product performance, safety and efficacy”. The concept of a design space has been introduced, which is defined as the multidimensional combination and interaction of input variables and process parameters that have been shown to provide assurance of quality (54). The underlying principles of QbD and risk management are contained in ICH Q8 (R2), Q9, and Q10 (50, 54–56).
A comprehensive case study of the application of QbD principles to the development of a mAb product (A-mAb) has recently been published that describes extensive use of scale-down models to gain process and product knowledge (50). However, significant data from scale-down models could be used to define the design space. In addition, data from triplicate studies performed at pilot or commercial scale using center-point conditions could ensure that product quality and process performance were not impacted by the process change.
Significant questions and challenges remain in implementing QbD. While there is agreement on the value of process and product understanding, there is often debate within most biopharmaceutical companies on the extent of QbD investment and underlying cost implications. Several technical hurdles need to be overcome to define methodologies to describe a design space and control strategy. Quality systems need to be developed that will enable the movement within a design space. Furthermore, as different regulatory agencies embrace QbD along different timelines, how will that affect a global submission? Finally how will QbD apply to more challenging large molecules such as vaccines? People involved in process development will be involved in the effort required to resolve many of these questions.
Conclusion
Biopharmaceutical development and manufacturing have evolved significantly in the past 25 years. Clinical and commercial pipelines have evolved from replacement proteins and other therapeutic proteins/hormones to mAbs. Technological advances in bioprocessing have led to tremendous increases in product and cell mass that in turn have posed several challenges to downstream process development of mAbs. Novel approaches to downstream processing, such as development of highly efficient platforms, HTS tools, and reduction in the number of unit operations have helped to address these challenges. In the future, mAbs may represent a smaller percentage of the pipelines and the challenges to downstream processing will likely have a different focus. Most product portfolios will likely include other complex biomolecules, such as conjugated proteins and vaccines, a variety of biosimilar protein therapeutics, and novel scaffolds such as fusion proteins, nanobodies, etc. Future expression systems may include microbial, yeast, and others in addition to high cell density mammalian systems. The diversity of scaffolds and expression systems will pose unique challenges for downstream development. Novel tools, approaches and/or platforms may need to be applied to enable rapid development. Furthermore, there is a need to develop paradigms to apply QbD not only to mAbs but also to other large molecules.
BERNARD N. VIOLAND is a research fellow, GLEN R. BOLTON is senior principal scientist, RICHARD S. WRIGHT is senior principal scientist, SHUJUN SUN is senior principal scientist, KHURRAM M. SUNASARA is senior principal scientist, KATHLEEN WATSON is associate director, JONATHAN L. COFFMAN is associate research fellow, CHRISTOPHER GALLO is associate research fellow, and RANGA GODAVARTI* is senior director, all from Pfizer. http://Ranga.Godavarti@pfizer.com/
References
1. C. Scott, Bioprocess Intl. 7 (10, Suppl.), (2009).
2. G. Walsh, Eur. J. Pharm. Biopharm. 55 (1), 3–10 (2003).
3. X. Wang et. al., mAbs. 1 (3), 254-267 (2009).
4. G. Jagschies et. al., BioPharm. Int. 19 (June Suppl.), 10–19 (2006).
5. J. R. Birch and A. J. Racher. Adv. Drug Deliv. Rev. 58 (5–6), 671-685 (2006).
6. B.D. Kelley, mAbs 1 (5), 1–10 (2009).
7. B.D. Kelley et.al., Biotechnol. Bioeng, 101 (3), 553–566 (2008).
8. J.L. Coffman, J.F. Kramarczyk, and B.D. Kelley, Biotechnol. Bioeng. 100 (4), 605–618 (2008).
9. B.D. Kelley et. al., Biotechnol. Bioeng. 100 (5), 950–963 (2008).
10. J. Glynn et. al., BioPharm. Int. 22 (October Suppl.), 15-19 (2009).
11. E.J. Suda, et. al., J. Chromatogr. A. 1216 (27), 5256–5264 (2009).
12. B. Kelley, Biotechnol. Prog. 23 (5), 995–1008 (2007).
13. G.R. Bolton, J. Basha, and D.P. LaCasse, Biotechnol. Prog. 26 (6), 1671–1677.
14. C. Winter, US patent 0237762 (2007).
15. G.R. Bolton, H. Araya, and A.W. Boesch, “Membrane Evaporation for Generating Highly Concentrated Protein Therapeutics” International Patent Application WO 2010/111378 A1.
16. M. Shamashkin et. al., Recovery of Biological Products XIV, (Lake Tahoe, CA 2010)
17. A.L. Nelson, E. Dhimolea, and J.M. Reichert, Nat. Rev. Drug. Discov. 9, 767–74 (2010).
18. G. Walsh, Nat. Biotechnol. 29 (9), 917–924 (2010).
19. C. Sheridan, Nat. Biotechnol. 28 (4), 307-310 (2010).
20. G. Walsh, BioPharma. Int. 23 (10), 1–8 (2009).
21. A.L. Nelson and J.M. Reichert, Nat. Biotechnol. 27 (4), 331–337 (2009).
22. M.D. Zolodz et. al., J. Chromatogr. A. 1217 (2), 225–234 (2010).
23. B.N. Violand. “Initial purification of inclusion bodies,” in Protein Purification Applications, S. Roe, Ed. (Oxford University Press, Oxford, UK, 2nd ed. 2001), pp. 19–27.
24. J.J. Buckley, S.D. Hoeltzli, and G.V. Johnson, Biotechnol. Appl. Biochem. 56 (1), 27–34 (2010).
25. V. Dechavanne et. al., Protein Expr. Purif. 75 (2), 192–203 (2011).
26. S.C. Alley, N.M. Okeley, and P.D. Senter, Curr. Opin. Chem. Biol. 14 (4), 529–537 (2010).
27. C.F. McDonagh et. al., Protein Eng. Des. Sel. 19 (7), 299–307 (2006).
28. J.R. Junutula et. al., Nat. Biotech. 26 (8), 92–-932 (2009).
29. D.P. Humphreys and L. Bowering, “Production of antibody Fab’ fragments in E. coli.” in Therapeutic Monoclonal Antibodies. Z. An, Ed. (John Wiley & Sons. Hoboken, NJ, 2009). pp 589–622.
30. R.E. Kontermann, Curr. Opin. Mol. Ther. 12 (2), 176–183 (2010).
31. C.E. Hagemeyer, et. al., Thromb Haemost. 101 (6) 1012-1019 (2009).
32. S.M. Kipriyanov., Methods Mol. Biol. 562, 205-214 (2009).
33. C.J. Fee. “Protein Conjugates Purification and characterization,” in PEGylated Protein Drugs: Basic Science and Clinical Applications, F.M. Veronese, Ed. (Die Deutsche Bibliothek Berlin Germany, 2009.) pp. 113–125.
34. T.M. Pabst et. al., J. Chromatogr. A. 1147 (2), 172–182 (2007).
35. F.V. Bockstaele, J.B. Holz, and R. Hilde, Curr. Opin. Invest. Drugs 10 (11), 1212–1224 (2009).
36. P.R. Brown, S.A Tobler, A.M. Wood, and A.W. Boesch, US patent application, SU2010/0172894 A1 (2010).
37. E. Burova and E. Ioffe. Gene Ther. 12 (suppl. 1s), S6–S17 (2005).
38. J.F. Wright. Gene Ther. 15 (11), 840–848 (2008).
39. R. Prylman, L. Schuerman. Expert Rev. Vaccines. 8 (11), 1479-1500 (2009).
40. T. Vesikari, et.al., The Pediatr. Infect. Dis. J. 28 (4), S66–S76 (2009).
41. FDA, Guidance for Industry: Evaluation of Combination Vaccines for Preventable Diseases: Production, Testing and Clinical Studies (Rockville, MD, April 1997).
42. G.T. Jennings and M.F. Bachmann, Annu. Rev. Pharmacol. Toxicol. 49, 303–326 (2009).
43. B. Keshab et. al., presentation at AIChE Annual Meeting (Salt Lake City, UT, 2009).
44. W. Waterfield, presentation at BioProcess International Conference & Exhibition (Providence, RI, 2010)
45. E. Shacter, presentation at American Chemical Society Conference (San Franscisco, 2010).
46. EMA CMHP, Guideline on Similar Biological Medicinal Products Containing Biotechnology-Derived Proteins as Active Substance: Non-Clinical and Clinical Issues (London, England, Feb. 2006).
47. C.K. Schneider and U. Kalinke, Nat. Biotechnol. 26 (9), 985-990 (2008).
48. G. Saberwal, Current Sci. 98 (12), 1575–1578 (2010).
49. R.L. Fahrner et. al., Biotechnol. Genet. Eng. Rev. 18, 301-327 (2001).
50. ISPE, “A-Mab: A Case Study in Bioprocess Development” 2009, http://www.ispe.org/PQLI_A_Mab_Case_Study_Version_2_1.pdf/, accessed Mar. 1, 2011.
51. P.J. Declerck et. al., Curr. Med. Res. Opin. 26 (5), 1219-1229 (2010).
52. S. Kozlowski, P. Swann. “Considerations for Biotechnology Product Quality by Design.” In Quality by Design for Biopharmaceuticals: Principles and Case Studies, A.S. Rathore and R Mhatre eds. (John Wiley & Sons, Inc., Hoboken, New Jersey, 2009) pp 9-27.
53. FDA, Notice of pilot program (Rockville MD, July 2008)
54. ICH, Q8(R2) Pharmaceutical Development (Geneva, August 2009).
55. ICH, Q10 Pharmaceutical Quality System (Geneva, May 2007).
56. ICH, Q9 Quality Risk Management (Geneva, November 2005).
Note: This article was first published in BioPharm International’s April 2011 supplement, and the original posting can be found here.


