By: Mark Mitchell
A practical roadmap in three parts that applies scientific knowledge, risk analysis, experimental data, and process monitoring throughout the three phases of the process validation lifecycle.
The most recent FDA (1) and International Conference on Harmonization (ICH) (2-4) guidance documents advocate a new paradigm of process validation based on process understanding and control of parameters and less on product testing. Consequently, the means of determining criticality has come under greater scrutiny. The FDA guidance points to a lifecycle approach to process validation (see Figure 1).
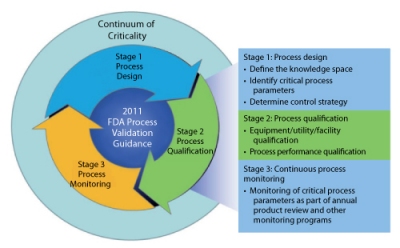
In Part I of this series, the author introduced the concept of continuum of criticality and applied it to the concepts of critical quality attributes (CQAs) and critical process parameters (CPPs). In the initial phase, the CQAs had their criticality risk level assigned according to the severity of risk to the patient. Applying a cause-and-effect matrix approach, the potential impact of each unit operation on the final product CQAs was assessed and each unit operation was thoroughly analyzed for its directly controllable inputs and outputs. Finally, a qualitative risk analysis or a formal failure mode effects and criticality analysis (FMECA) was conducted for each of the identified process parameters. The purpose of this assessment is to provide a focus for the downstream process characterization work required to complete process validation Stage 1 (process design).
This initial risk assessment is performed prior to the baseline characterization work and can be used as the primary means of determining the criticality of process parameters under the following conditions:
- When a platform process that possesses similar properties and process to another commercial product (e.g., new strength or new dosage form)
- When there is a significant body of published data on the process
- When experimental studies and commercial data are available, such as when the process validation lifecycle is applied to a legacy product to substantiate the initial assessment.
In these cases, this initial assessment can be further bolstered through the addition of an uncertainty component to the traditional risk score. For example, a high-risk critical parameter with low uncertainty (due to substantial supporting data) may not require further study, but a medium-risk parameter with high uncertainty may require further experimentation to quantify the risk to product performance.
The challenge facing most organizations is how to effectively evaluate the impact of potentially hundreds of process parameters on product performance to determine what is truly critical. Few companies have the time or resources to design experimental studies around all potentially critical process parameters. The initial risk assessment provides a screening tool to sort out the parameters that have low or no risk.
Design space and design of experiments
The goal is to increase process knowledge by providing a mechanistic understanding of the relationship between process parameters, raw material attributes, and CQAs. This is defined as both the demonstration of impact and the quantification of the contribution of each parameter to the product’s performance. Through this exercise, it will be possible to identify the process design space. The ICH guidance defines three elements–knowledge space, design space, and control space–to establish a process understanding (see Figure 2) (2).
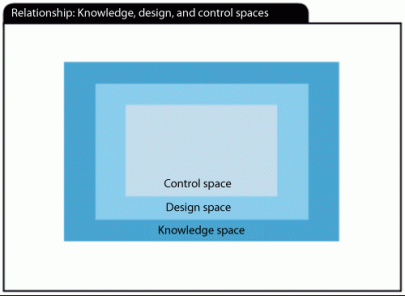
ICH Q8 defines design space as, “The multidimensional combination and interaction of input variables (e.g., material attributes) and process parameters that have been demonstrated to provide assurance of quality.”
The design space is part of an enhanced process development approach referred to as quality by design (QbD). Prior to QbD, pharmaceutical development did not require the establishment of functional relationships between CPPs and CQAs. Consequently, process characterization experiments were primarily univariate (one factor at a time [OFAT]), showing that, for a given range of a process parameter (referred to as proven acceptable range or PAR), the CQAs meet acceptance criteria. While univariate experiments can provide some limited knowledge, a compilation of OFAT studies cannot typically define a design space because it cannot substantiate the importance or contribution of the parameter to the product CQA being evaluated. To do this, multivariate studies must be performed to account for the complexities of interactions when several CPPs vary across their control ranges.
Design spaces can be developed for each unit operation or across several or all unit operations. Although it may be simpler to develop for each unit operation, downstream unit operations may need to be included to sample and test the appropriate CQAs. For example, to perform a multivariate study on a fermentation unit operation, additional processing through cell lysis and purification unit operations is needed so that CQAs may be sampled and tested. The challenge faced by most development programs is how to efficiently and cost-effectively derive maximum process understanding in the fewest number of studies. To do this, a staged approach using multiple studies is most efficient.
A staged design of experiment approach
The following is an example of a simple staged design of experiment (DOE) approach. More complex DOE designs and strategies may be required, but these designs are typical:
- Screening (fractional factorial, Plackett-Burman). To identify or screen out process parameters that have no significant impact on a CQA. Screening designs can test main (individual impact and contribution) effects of each parameter being evaluated.
- Refining (full factorial). Having dropped out parameters, which do not impact the product CQAs, the refining step tests both main effects and interactions between the remaining parameters and generates first-order (linear) relationships between process parameters and CQAs. The criticality level of a CPP is determined from the quantitative impact on the CQA shown in the modeled relationship.
- Optimization (central composite, Box-Behnken). To generate response surfaces and illustrate second-order (quadratic) relationships between process parameters and CQA. This analysis allows optimal set points for the design space or control space to be identified to target desired CQA (or performance attributes) values.
The DOE design assists in determining which parameters are studied and what set point value is used for each experimental run. The initial risk assessment, using prior knowledge and scientific principles, provides an expected relationship as to which CQAs and their related in-process controls will be affected by the given process parameters. Although the focus is on quality impact, process performance attributes (no quality impact) should also be sampled and measured as appropriate. This step is especially important during the optimization stage because a trade-off may be required in terms of optimizing quality and performance attributes.
For process validation Stage 1 process characterization studies, analytical methods for measuring CQAs may not yet be fully validated, but still, must be scientifically sound.
The level of accuracy and precision of the analytical method or measurement system must be well understood because they directly impact the quantitative decision process when interpreting study results early in the process-design stage. Techniques of measurement system analysis such as Gage repeatability and reproducibility (Gage R&R) studies are recommended because they provide information on the variability of the measurement system. The Gage R&R study provides a quantitative measurement of the measurement tools contribution to variation for any measurement made. Typically, a percent contribution from R&R variability must be < 20% and demonstrate at least five distinct categories for the method result to be meaningful. The distinct categories are the number of discernable groups of measurements that can reliably span the range of the CQA.
Including replicate runs in addition to the study experimental design provides crucial data for estimating the underlying variability of the study. This is because, during each run, small unmeasured and uncontrolled variations always occur and may influence the result. Two otherwise identically configured runs may produce slightly different responses due to changes in environment, equipment, measurement, sampling, and operators, among others. Even deliberately fixed parameters (those not under study) may not be exactly identical from run to run. Together, these are called “noise” factors and are important in discerning true responses (i.e., “signal”) caused by the changing parameters from the inherent variability. Differences between sets of replicate runs allow for the quantification of this variability. Large changes in the responses between replicates may indicate either an unstable experimental platform (such as poor run-to-run control) or that a low-risk CPP or non-CPP, may have a higher impact on the CQAs than originally assessed.
Where a raw material has a critical material attribute (CMA) of medium-risk to high-risk impact to CQAs, it should be included as a parameter of the study where possible. Multiple lots or lots with extreme variation in CMAs may not always be available during early development or characterization studies. This limitation frequently is one of the primary drivers of establishing the continuous process verification (CPV) program in Stage 3 to monitor the future impact from this raw material variation. For large studies, multiple lots of raw materials may be required. Consideration should be given to either proportional mixing of the raw material lots for each run or use of a statistical technique called blocking, which incorporates change of material lots into the experimental design.
In each design, the choice must not only be made on the number of parameters to be studied, but at how many levels (i.e., set points within the range) and how many times a particular set of conditions is repeated (replication).
The number of levels is related to the mathematical relationship between the parameters and the CQA measured (e.g., two levels for linear or three for quadratic). For screening designs, it is typical to use only two levels (minimum and maximum of the range); for these designs, any known non-linear relationship may have to be mathematically transformed. For refining design, center points (midpoint of ranges for all parameters) are added to estimate variability and to detect potential curvature.
Because of the cost involved or availability of API, it is not possible to perform all experimental studies at commercial scale (such as with fermentors of 5000 to 25,000 L), hence, most biotech process development programs rely heavily on modeling the process at smaller or intermediate scale. Some process parameters may be independent of the scale or may have simple models to account for scale changes. Scale itself may be considered a parameter. Establishing similar run conditions at multiple scales is an important consideration when trying to qualify the comparability between full-scale and small-scale experiments. Substantial prior experience with scaling particular unit operations may provide key information such as dimensionless parameter groups and scaling equations.
Areas considered for experimental scale include, but are not limited to:
- Aspect ratios of bioreactors and mixing tanks
- Impeller number, size, and location
- Aeration method and effectiveness of oxygen transfer
- Location of addition ports and effect on mass transport and uniformity
- Temperature control and heat-transfer surface area
- Location of instrument sensors and control-loop tuning parameters.