By: Mark Mitchell
Cause-and-effect matrix
Once risk levels have been assigned to the CQAs, the next activity is to begin to relate which parts of the process have impact on these attributes. This cause-and-effect analysis breaks the process into its unit operations and conveys its impact on the CQAs. An example of the cause-and-effect matrix for a biologic is given in Table I.
Table I: Example of cause and effect matrix for a biologic. H = high, M = medium, L = low.
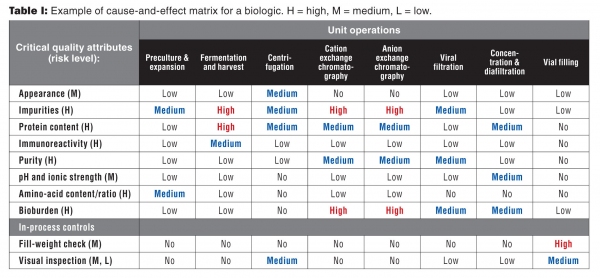
In addition to the matrix, it is important to document the justification for these decisions as part of this analysis. For example, the parameters of the cation- and anion-exchange chromatography processes are expected to have a high impact on impurities because they are designed to remove impurities of different ionic charge than the desired product. With the knowledge of which unit operations have impact on particular CQAs, it is now possible to analyze each process’ inputs and outputs to determine how process parameters affect the CQAs.
Input and output process variables
Each unit operation has both input and output process variables. Process parameters signify process inputs that are directly controllable and can theoretically impact CQAs. Process outputs that are not directly controllable are attributes. When the attribute ensures product quality, it is a CQA. The output of one unit operation can also be the input of the next unit operation. These parameters and attribute designations and their justification should be documented in either a formal process description document, or process flow diagram/drawing. This documentation should also include the scale of each unit operation, equipment and materials required, sampling/monitoring points, test methods, and relevant processing times and storage times/conditions.
The intent of assessing process parameters is to determine how they affect the process variation of CQAs along with their control strategy. Each company should clearly document their methodology for defining and assessing parameters. The following may be considered in making that assessment:
- Raw material attributes are outputs of the release of materials. Critical material attributes (CMA) should be considered along with CPPs as impacting process variability.
- Fixed parameters such as equipment scale, equipment setup, pre-programmed recipes should be documented but are assessed as either low or non-critical.
- Parameters for sterilization processes and cleaning process and the preparation of process intermediates can be included in the primary process assessment. Alternatively, they can be treated as independent processes with their own process parameters, quality attributes, criticality assessments, and process validation.
- Calibration and standardization setting for equipment and instruments are usually not included as process parameters.
- Formulation recipes can be considered fixed parameters (low or not critical); these parameters generally have relatively tight limits, which are justified during formulation development. Such a parameter, which does not vary, cannot impact process variability. An exception to this rule is the case where operators must calculate a quantity based on a variable input such as biological activity; this variable process parameter may lead to process variation.
- Holding/storage times and conditions where no processing occurs should be qualified to show little to no impact on the product. These should be documented and, if these factors are included as process parameters, they are considered low or non-critical.
- Environmental conditions during process (room temperature, humidity), such as holding times, are to have set limits so that they have little to no impact to the process. Process-specific environmental conditions such as cleanrooms, cold rooms, and dry rooms are included as process parameters because they are monitored to ensure product quality.
When a process parameter is determined to be non-critical either by process knowledge or by process study, companies may choose to further designate the parameter as a key performance parameter if that parameter impacts a process performance attribute.
From knowledge to risks
Once each unit operation is related to CQAs through a cause-and-effect matrix and the process parameters and attributes are documented, an initial risk assessment to determine the potential impact of each process parameter is performed. Prior to process characterization experiments, this risk assessment may be more high level using primarily prior knowledge and scientific principles. However, a more formal FEMCA may also be considered.
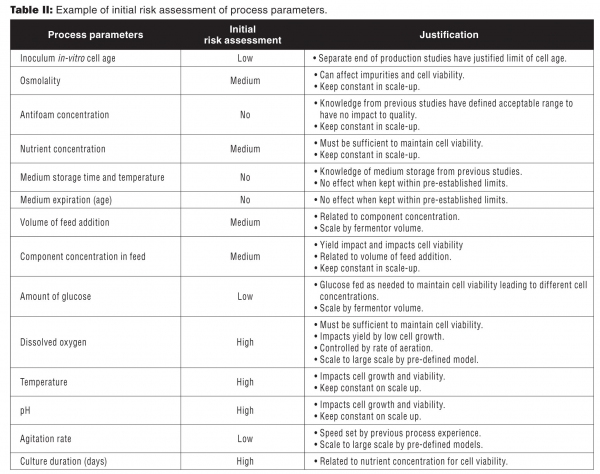
Table II is an example of an initial risk assessment for a single unit operation. Included in the justification is the expected relationship with CQAs and how the parameter may be influenced during scale-up. Fixed parameters are set to non-critical as they do not impact process variability. For the initial process characterization experiments, process parameters with medium to high impact will be included.
Table II: Example of initial risk assessment of process parameters
In Part I of this series, the author looked at criticality as a continuum to apply risk analysis during process design, and to relate process unit operations to quality attributes using a cause-and-effect matrix.
In Part II, the continuum of criticality for parameter and attributes will be used to design process characterization studies using DOE. From the initial risk assessment of critical parameters, experimental data from formal studies will confirm the criticality assignment—critical or not—and help to assess the level of impact to CQAs.
References
1. FDA, Guidance for Industry, Process Validation: General Principles and Practices, Revision 1 (Rockville, MD, January 2011).
2. ICH, Q8(R2) Harmonized Tripartite Guideline, Pharmaceutical Development, Step 4 version (August 2009).
3. ICH, Q9 Harmonized Tripartite Guideline, Quality Risk Management (June 2006).
4. ICH, Q10, Harmonized Tripartite Guideline, Pharmaceutical Quality System (April 2009).
5. ICH, ICH Quality Implementation Working Group Points to Consider (R2), ICH-Endorsed Guide for ICH Q8/Q9/Q10 Implementation (6 December 2011).
6. ISO/IEC Guide 51: Safety Aspects-Guidelines for their inclusion in standards, 2nd ed. (1999).
7. IEC 60812, Analysis Techniques for System Reliability-Procedure for Failure Mode and Effects Analysis (FMEA), Edition 2.0 (January 2006).
8. ISPE, Product Quality Lifecycle Initiative (PQLI) Good Practice Guide, Overview of Product Design, Development, and Realization: A Science- and Risk-Based Approach to Implementation (October 2010).
9. ISPE, Product Quality Lifecycle Initiative (PQLI) Good Practice Guide, Part 1-Product Realization using QbD, Concepts and Principles (2011).
10. Parenteral Drug Association, Technical Report 60, Process Validation: A Lifecycle Approach (2013).


